While playing with his sister, 5-year-old Harry Eastlack broke his leg. He was taken to the hospital to set the fracture, but it never healed properly. Then, at age 10, Harry’s knee and hip joints stiffened. Strange lumps formed in his thigh muscle – lumps of bone. When surgeons removed these lumps, new bone growths appeared in their place, larger and more extensive than before. By age 20, all of the vertebrae in Harry’s spine had fused into one piece and his most of his back muscles had ossified into boney plates. Harry’s muscles, tendons, and ligaments were slowly turning into bone [1]. He suffered from an extremely rare disorder, called fibrodysplasia ossificans progressiva or FOP, caused by a mutation in this week’s gene of interest: activin receptor-like kinase-2 or ALK-2.
FOP is the body’s repair mechanism gone awry. Normally injuries are repaired by regenerating wounded tissue. Tear a muscle, new muscle regrows in its place. But people with FOP ‘heal’ connective tissue (muscle, tendons, and ligaments) by converting the wounded tissue into bone, spontaneously or upon injury [2]. Even small wounds, like a needle prick from an injection, can form tiny bone spurs under the skin. FOP sufferers dread bumps or falls – a car crash would be a nightmare – because any injury might be ‘healed’ by bone growth that permanently locks the affected joints in place. Even without injury, the disease progresses through the body from top to bottom, starting at the neck, then shoulders, arms, around the ribs, and finally the legs and feet [3]. Over time, each joint turns to bone, locking limbs at awkward angles and slowly converting a person into a living statue.
Again, bone formed in FOP isn’t new bone growth, but conversion of normal tissue into bone. While that sounds pretty disturbing, it’s not a completely abnormal process, but takes its roots in normal bone development.
As I mentioned in an earlier post, bone is a living tissue, with the potential to grow, adapt, and heal itself. There are two ways bones form [4]. One, intramembranous ossification, forms thin plates of bone like those found in the skull. The other, endochondral ossification, starts with a template of cartilage that’s converted into bone. This is how most of the bones in the skeleton are formed. A temporary cartilage skeleton develops and is replaced with bone, converting one type of tissue into another. Exactly like FOP.
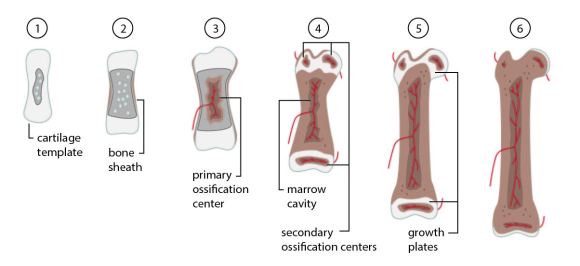
1) A cartilage template forms. Chondrocytes (cartilage-forming cells) in the center start to die off leaving small cavities. 2) Cells around the shaft of the cartilage template convert into osteoblasts (bone-forming cells). The osteoblasts create a sheath of bone around the template. 3) Blood vessels invade the center of the template. Fibroblasts (wound healing cells) migrate in through the blood vessels and convert into osteoblasts. These newly-converted osteoblasts produce bone, forming the primary ossification center. 4) The marrow cavity forms in the center. Secondary ossification centers develop at each end. 5) The left over cartilage between ossification centers forms the growth plate, allowing the bone to grow and lengthen through adolescence. 6) The growth plate is replaced with bone in adulthood. Cartilage remains only at the joints.
Bone growth in controlled by a set of growth factors called bone morphogenic proteins or BMPs [5]. Similar to the receptor Her2, BMP signals its bone-growth message through a set of cellular receptors that sit on the outside of the cell. One of those receptors is ALK-2. When BMPs interact with ALK-2, it sends pro-bone signals into the cell.

Harry Eastlack’s Skeleton on display at the Mutter Museum. Source: Joh-co / Wikimedia Commons
In FOP, a single amino acid in ALK-2 is switched with another [6]. This mutation leaves ALK-2 in the ‘on’ position, allowing it to send pro-bone signals even without the presence of BMP growth hormones. With this signal always on, it takes very little for cells in the body to undergo the transition similar to that found in endochondral ossification – and convert normal tissues into bone.
Much of what we know about FOP is thanks to Harry Eastlack and his skeleton, on display at The Mütter Museum of The College of Physicians in Philadelphia [1]. By the end of his life, Harry’s skeleton had fused into one solid piece. He could only move his lips. On his death bed, just a few days shy of his 40th birthday, he asked to donate his body to science, hoping it would help scientists understand this horrifying disorder. The bridges and plates wrapping his skeleton like papier-mâché pointed to uncontrolled endochondral ossification. This valuable hint helped identify the ALK-2 mutation which might eventually lead to a cure.
References
[1] “Bone, A Masterpiece of Elastic Strength”. New York Times. 27 April 2009.
[2] van Dinther, M.; Visser, N.; de Gorter, D.J.; Doorn, J.; Goumans, M.J.; de Boer, J.; ten Dijke, P. (2011) “ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation”. J Bone Mineral Res. 25 (6): 1208-15. PMID 19929436.
[3] “FOP Symptoms”. IFOPA. 2009.
[4] Gilbert SF (2000) “Osteogenesis: The Development of Bones”. Developmental Biology. 6th edition. Sunderland (MA): Sinauer Associates.
[5] Chen, D.; Zhao, M.; Mundy, G.R. (2004) “Bone morphogenetic proteins”. Growth Factors 22(4): 233-41. PMID 15621726
[6] Shore, E.M.; Xu, M.; Feldman, G.J.; et al. (2006). “A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva”. Nat. Genet. 38(5): 525–27. PMID 16642017.



 In Ian Fleming’s novels, all 00 agents are issued cyanide capsules in case of enemy capture, but James Bond threw his away. What a dummy. Sure, Bond always escapes, but why risk it? The rest of his peers understood their orders and were ready to die to keep their country’s secrets. Cyanide pills aren’t mere tropes either, but a real spy tool – a gruesome last resort to keep information out of enemy hands [1]. A lethal dose of cyanide kills in minutes – so effective because it blocks the body’s ability to make energy, via this week’s gene of interest: ATP synthase gamma subunit (ATP5C1).
In Ian Fleming’s novels, all 00 agents are issued cyanide capsules in case of enemy capture, but James Bond threw his away. What a dummy. Sure, Bond always escapes, but why risk it? The rest of his peers understood their orders and were ready to die to keep their country’s secrets. Cyanide pills aren’t mere tropes either, but a real spy tool – a gruesome last resort to keep information out of enemy hands [1]. A lethal dose of cyanide kills in minutes – so effective because it blocks the body’s ability to make energy, via this week’s gene of interest: ATP synthase gamma subunit (ATP5C1).

 People can’t live without vitamin C, but everyone’s heard a story about that one college student who tried to live off of nothing but chips and ramen [1]. And were it not for a mutation a few millions of years ago, the story wouldn’t end in the clinic with a diagnosis of scurvy. We could synthesize all the vitamin C we need with a few repairs to this week’s gene of interest: L-gulono-γ-lactone oxidase or GLO.
People can’t live without vitamin C, but everyone’s heard a story about that one college student who tried to live off of nothing but chips and ramen [1]. And were it not for a mutation a few millions of years ago, the story wouldn’t end in the clinic with a diagnosis of scurvy. We could synthesize all the vitamin C we need with a few repairs to this week’s gene of interest: L-gulono-γ-lactone oxidase or GLO.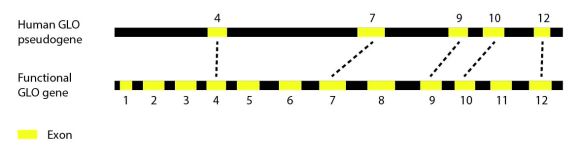
 So that’s why you should eat your fruits and veggies – because evolution has failed you. But considering the American diet, it’s probably a good thing we can’t make our own vitamin C. If not for a horrifying death by scurvy, too many people would opt to avoid anything green for tasty, tasty junk food.
So that’s why you should eat your fruits and veggies – because evolution has failed you. But considering the American diet, it’s probably a good thing we can’t make our own vitamin C. If not for a horrifying death by scurvy, too many people would opt to avoid anything green for tasty, tasty junk food.







 NOTCH is a cell-cell receptor, which means it sits on the surface of the cell (like other
NOTCH is a cell-cell receptor, which means it sits on the surface of the cell (like other 



