Rare genetic disorders are hard to study because they’re rare. With public awareness almost nil, funding doesn’t come easy so treatments develop slowly. But a few ‘lucky’ disorders present in notable patients, boosting awareness and jump-starting research. Hemophilia is one of the lucky few, and it’s caused by a mutation in this week’s gene of interest: coagulation factor IX or F9.

Prince Leopold, Duke of Albany
‘Royal’ hemophilia first appeared in Prince Leopold, the son of Britain’s Queen Victoria. A delicate boy, Leopold bruised and hemorrhaged at the slightest bump or scratch, and these wounds would take longer than normal to heal – a result of poor coagulation [1]. Little was known about hemophilia at the time, but physicians tried everything to treat the disease. While some treatments made sense – like applying ice and compressing the wound to slow bleeding – others were a little more creative – like treating the wound with lime, hydrogen peroxide, or diluted snake venom. If bleeding was severe, they would also transfuse hemophiliacs with the blood of a relative – a potential disaster if their blood was incompatible [2]. Ultimately, hemophiliacs like Leopold were destined to live short, sheltered lives as they could bleed to death from nearly any injury.
Queen Victoria tried to cover up Leopold’s condition, likely swearing his physicians to secrecy to protect the dignity of the bloodline [3]. While understanding of the disease was still limited, researchers had noticed it was hereditary. If word of Leopold’s hemophilia got out, Victoria’s other children might have trouble marrying into other royal families. She kept it a secret as best she could and hoped the disease was isolated to Leopold. She was wrong.
Historians now believe Queen Victoria was the source of the hemophilia mutation that spread through European royalty [3]. We now know that two of her daughters were carriers and passed the mutation on to other royal families in Spain, Germany, and Russia [1].
Carriers can have the mutation, but never suffer from hemophilia. Normally, you need two bad copies of a gene (one on each set of chromosomes) for a genetic disorder to present. A single bad copy makes you a carrier – you stay healthy because of your one working copy, but you might pass on the bad copy to your children. But hemophilia is X-linked – F9 is located on the ‘X’ chromosome. Girls have two ‘X’ chromosomes. Boys have only one ‘X’ and one ‘Y’. Therefore, a girl can inherit a single bad copy of F9 and have no issue, but if a boy inherits a single bad copy, he’ll develop hemophilia [4].
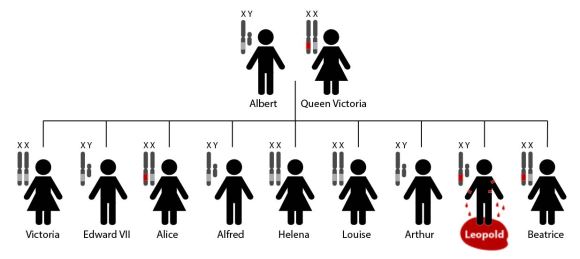
Queen Victoria’s family tree: A light gray band indicates a normal copy of the F9 gene. A red band indicates a mutated copy of F9. Alice and Beatrice were both carriers of the mutated F9 gene while Leopold, with his single faulty copy of F9, developed hemophilia [1].
References
[1] “Hemophilia: ‘The Royal Disease'”. National for Case Study Teaching in Science. 20 September 2003.
[2] “History of Bleeding Disorders”. National Hemophilia Foundation.
[3] “A Royal Shame: Prince Leopold’s Hemophilia and Its Effect on Medical Research”. Dartmouth Undergraduate Journal of Science. 22 May 2009.
[4] Rogaev, E.I.; Grigorenko, A.P.; Faskhutdinova, G.; Kittler, E.L.; Moliaka, Y.K. (2009). “Genotype analysis identifies the cause of the ‘royal disease'”. Science. 326 (5954): 817. PMID 19815722







 NOTCH is a cell-cell receptor, which means it sits on the surface of the cell (like other
NOTCH is a cell-cell receptor, which means it sits on the surface of the cell (like other 



